pORTMAGE & pSEVAMAGE Protocol
This page aims to help genome-engineering endeavors using the pORTMAGE or pSEVAMAGE plasmid sets, with a detailed description of our tried-and-tested experimental procedure.
Genome Engineering has opened a new avenue of research through the precise editing of cellular DNA that is orders of magnitude faster than natural mutational processes. It allows us to rationally construct mutations and mutational combinations and thereby study long-term evolutionary processes within laboratory timescales.
Evolutionary Genome Engineering (Pál, Papp, and Pósfai 2014), a new field in evolutionary biology unlocks far-reaching opportunities in fundamental- and applied research. From the directed evolution of novel phenotypes, metabolic functions, and enzymatic activities to answering fundamental problems in genetics, e.g. the conservation of mutational effects and epistasis, it has immediate applications. Moreover, with its ability to recapitulate evolutionary innovations in a massively parallel manner and thereby understand the mechanism behind them, it holds out hope to forecast evolution.
Single-stranded DNA oligonucleotide (oligo)-mediated allelic exchange (Ellis et al. 2001) (Costantino and Court 2003) (Aparicio et al. 2016) (Van Kessel and Hatfull 2008) (Pijkeren and Britton 2014) (recombineering) is an especially powerful tool for genome editing in bacteria. It is based on the ssDNA annealing protein (Beta) of the λ-Red recombination machinery and catalyses efficient annealing of short ssDNA oligos to the lagging strand of open replication forks. Small modifications, encoded within single oligonucleotides, can be repeatedly introduced without selection to achieve targeted reprogramming at multiple loci of the host genome. This approach, coupled with rational DNA design and massively-parallel DNA manufacturing, has become a fundamental tool in genome engineering.
Recombineering can be multiplexed, that is multiple oligos are capable of modifying their genomic targets simultaneously. These modifications can range from single mutations (e.g. individual SNVs) to large-scale genome refactoring. The straightforward computational design and cost-effective synthesis of the mutagenizing DNA strands (90 nucleotide long DNA oligonucleotides) made recombineering a fundamental tool for Evolutionary Genome Engineering. This technique – called Multiplex Automated Genome Engineering (MAGE) (Wang et al. 2009) – is currently the most versatile and high-throughput approach for truly genome-scale editing, allowing for the continuous generation of novel genetic variants within a population.
Reference: Nyerges Á, Csörgő B, Nagy I, Bálint B, Bihari P, Lázár V, Pál C, et al. 2016. A highly precise and portable genome engineering method allows comparison of mutational effects acrossbacterial species. Proceedings of the National Academy of Sciences of the United States of America
Using a dominant negative mutant protein of the methyl-directed mismatch repair (MMR) system, the plasmids allow transient suppression of DNA repair in enterobacterial species, which is necessary for efficient oligonucleotide integration. By integrating all necessary components into the Standard European Vector Architecture format, the pSEVAMAGE vector set facilitates rapid MAGE applications in a diverse set of enterobacterial species.
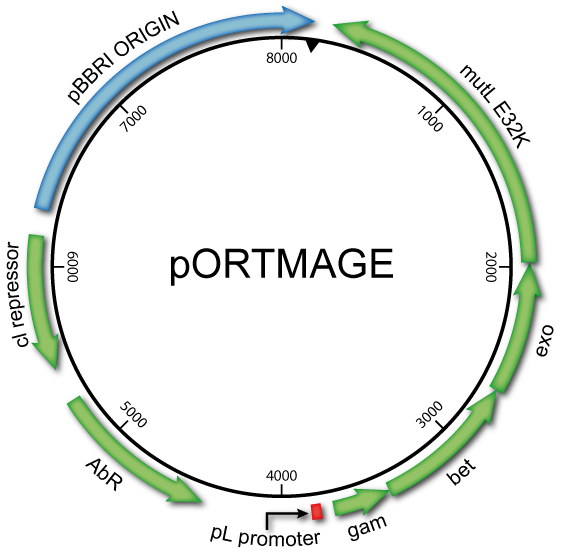
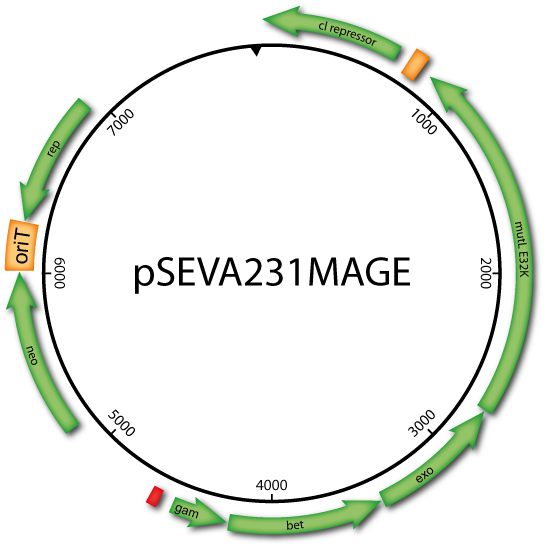
pORTMAGE and pSEVAMAGE allow efficient modification of multiple loci without prior modification of the host genome. Due to the conserved nature of the bacterial MMR system, they simultaneously allow genome editing and mutant library generation in a wide range of biotechnologically and clinically relevant enterobacterial species.
pORTMAGE and pSEVAMAGE plasmids are easily incorporated into the standard workflow of MAGE (for background information and detailed design of MAGE experiments see References 1-6)
and both plasmids are available via the Standard European Vector Architecture
(SEVA http://www.cnb.csic.es/index.php/en/);
Addgene https://www.addgene.org/Csaba_Pal/,
and directly from our laboratory (For direct strain request and further questions, please contact: nyerges.akos
Experimental workflow for pORTMAGE
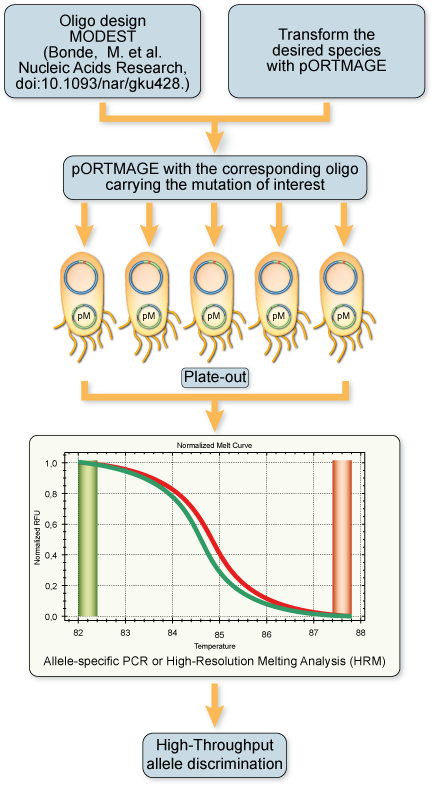
Cells and reagents:
- Target species, transformed with the corresponding pORTMAGE, pSEVA221MAGE or pSEVA231MAGE plasmid:
| Plasmids | PlasmidMarker gene | Note | Sequence |
|---|---|---|---|
| pORTMAGE2 | AmpR | derived from pSIM8 (Datta et al.) | download |
| pORTMAGE3 | KanR | KanR derivative of pORTMAGE2 | download |
| pORTMAGE4 | CmR | CmR derivative of pORTMAGE3 | download |
| pSEVA221MAGE | KanR | pSEVA221 based derivative of pORTMAGE3 | |
| pSEVA231MAGE | KanR | pSEVA231 based derivative of pORTMAGE3 | download |
- Oligonucleotides for MAGE (for design: see Reference 1, 2, 4 and 6):
Ordered with standard desalting from IDT and designed according to the general guideline Gallagher, et al. (2014) : 90 nt length, 2 phosphorotioate bonds at each termini, minimized secondary structure (dG >-12 kcal/mol) and should be screened for mistargets on the corresponding genome to avoid false hybridization.
- Chilled water (+4°C) for electrocompetent cell preparation
- Luria-Bertani-Lennox (LBL) broth: 10 g of tryptone, 5 g of yeast extract, 5 g of sodium chloride
- Antibiotic-supplemented LBL broth and agar plates with the corresponding antibiotic
- Terrific-broth (TB): yeast extract 24 g, tryptone 12 g, K2HPO4 9.4 g, KH2PO4 2 g per 1L of water
LABWARE:
- Shaking water bath (42°C)
- Shaking incubator for flasks (30°C)
- BioRad MicroPulser or BTX (Harvard Apparatus) CM-630 Exponential Decay Wave Electroporation System
- Refrigerated centrifuge with swing-out rotor
- Electroporation cuvettes with 1 mm (VWR Signature Electroporation Cuvette) gap
- 150 ml glass flask
MAGE cycling protocol:
- Start with a freshly streaked, individual colony from an LBL agar + antibiotic plate (grown at 30°C, overnight)
- Inoculate into a glass flask, containing 2 ml LBL aliquot, supplemented with the corresponding antibiotic
- Incubate in a shaking incubator at 250 rpm at 30 °C overnight
- From the stationary phase culture, dilute cells 100-fold in LBL, supplemented with the corresponding antibiotic in a 150 ml glass flask
- Incubate in a shaking incubator at 250 rpm at 30 °C
- Upon reaching OD550 0.55-0.65, transfer cells to the 42°C shaking water bath to induce λ Red protein expression for 15 min at 250 rpm
- Place cells immediately on ice and chill for at least 5 min
- Prepare electrocompetent cells by washing and pelleting them twice in 10 ml ice-cold dH2O at 3800 rpm for 7 min in a refrigerated centrifuge with swing-out rotor at 4 °C
- Suspend cells in 160 μl dH2O and keep on ice until electroporation
- For electroporation, mix 40 μl of competent cells with 1 μl 100 μM MAGE oligo (resuspended in TE buffer)
- Electroporate cells in a pre-chilled 1 mm gap cuvette (e.g. VWR Signature Electroporation Cuvette) using a BioRad MicroPulser or BTX (Harvard Apparatus) CM-630 Exponential Decay Wave Electroporation System (parameters: 1800 V, 25 μF, 200 Ω)
- Immediately after electroporation, add 1 ml room temperature TB medium
- Transfer cell suspension immediately to 4 ml prewarmed (30 °C) TB medium, in a 150 ml glass flask
- Allow to recover for 60 min at 30°C at 250rpm
- Add 5 ml room-temperature LBL medium supplemented with the corresponding antibiotic
- Grow cells at 30 °C until mid-logarithmic state (OD550 0.55-0.65) at 250 rpm
- At this point, cells could be either subjected to additional MAGE cycles or assayed for phenotype and genotype analysis
References:
Wang, Harris H., Farren J. Isaacs, Peter A. Carr, Zachary Z. Sun, George Xu, Craig R. Forest, and George M. Church. (2009)
Programming Cells by Multiplex Genome Engineering and Accelerated Evolution.
Nature 460 (7257): 894–98.
Gallagher, Ryan R., Zhe Li, Aaron O. Lewis, and Farren J. Isaacs. (2014)
Rapid Editing and Evolution of Bacterial Genomes Using Libraries of Synthetic DNA.
Nature Protocols 9 (10): 2301–16.
Carr, Peter A., Harris H. Wang, Bram Sterling, Farren J. Isaacs, Marc J. Lajoie, George Xu, George M. Church, and Joseph M. Jacobson. (2012)
Enhanced Multiplex Genome Engineering through Co-Operative Oligonucleotide Co-Selection.
Nucleic Acids Research 40 (17): e132–e132.
Bonde, Mads T., Mads V. Anderson, Annika I. N. Wallin, Harris H. Wang, and Morten O. A. Sommer. (2014)
MODEST: A Web-Based Design Tool for Oligonucleotide-Mediated Genome Engineering and Recombineering.
Nucleic Acids Research, May, gku428. doi:10.1093/nar/gku428
Nyerges, Á., Csörgő, B., Nagy, I., Latinovics, D., Szamecz, B., Pósfai, G, Pál, C. (2014)
Conditional DNA repair mutants enable highly precise genome engineering
Nucleic Acids Research, 2014,1–10 doi:10.1093/nar/gku105
Silva-Rocha, Rafael, Esteban Martínez-García, Belén Calles, Max Chavarría, Alejandro Arce- Rodríguez, Aitor de las Heras, A. David Páez-Espino, et al. (2013)
The Standard European Vector Architecture (SEVA): A Coherent Platform for the Analysis and Deployment of Complex Prokaryotic Phenotypes.
Nucleic Acids Research 41 (D1): D666–75. doi:10.1093/nar/gks1119
Further information:
Standard European Vector Architecture (SEVA)
DTU – MODEST site